2021 09 06 生物信息学从零开始学 5 蛋白质三级结构预测
森言森语
昔日龌龊不足夸,今朝放荡思无涯。
春风得意马蹄疾,一日看尽长安花。
——登科后 (唐)孟郊
接下来的几天写一下关于分子对接的专题,今天的推文是导入,先了解一下基本的知识点。
□ 计算方法预测蛋白质三级结构
- 从头计算法(ab initio)
- 同源建模法(homolog modeling)
- 穿线法(threading)
- 综合法(ensemble method)
1 homolog modeling: SWISS-MODEL
原理:相似的氨基酸序列对应着相似的蛋白质结构 步骤:
- 找到与目标序列同源的已知结构作为模板(目标序列与模板序列之间的一致度要≥30%)
- 为目标序列与模板序列(可以多条)创建序列比对。通常比对软件自动创建的序列比对还需要进一步人工矫正。
- 根据第二步创建的序列比对,用同源建模软件预测结构模型 - 评估模型质量,并根据评估结果重复以上过程,直至模型质量合格。
https://swissmodel.expasy.org/
预测效果 如果目标序列与模板序列一致度极高,那么同源建模法是最准确的方法。
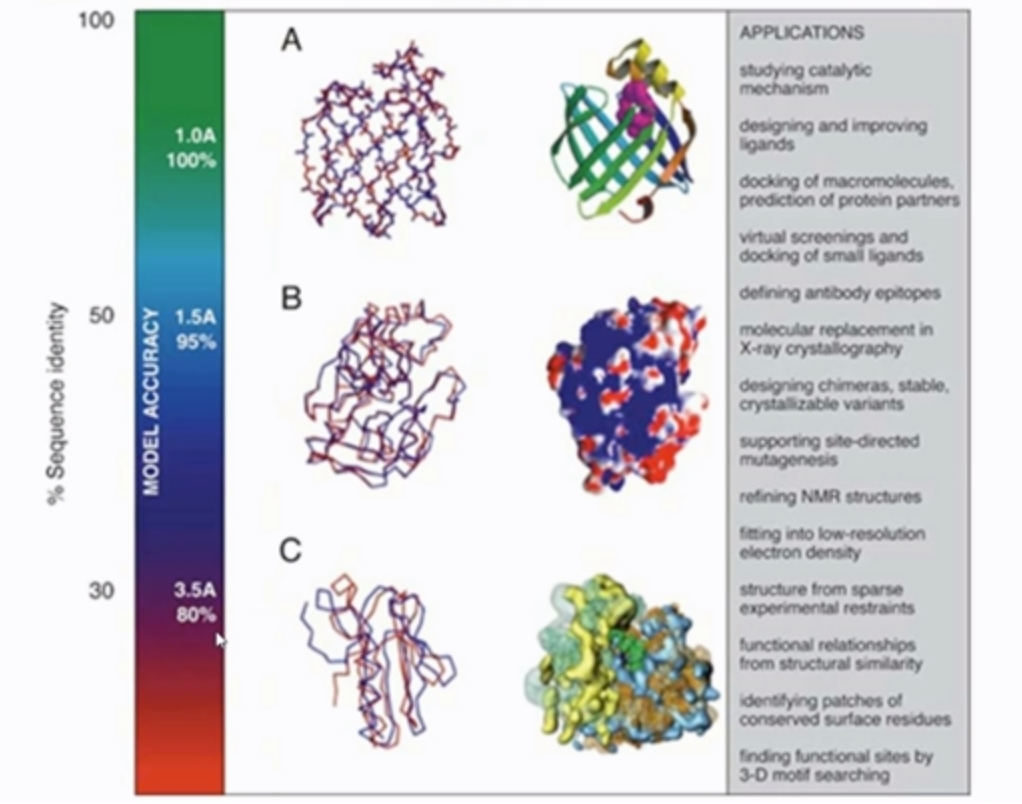
特例情况 虽然序列一致度达到很高水平,但是结构却并不相同。
2 threading:I-TASSER
原理:不相似的氨基酸序列也可以对应着相似的蛋白质结构
https://zhanglab.ccmb.med.umich.edu/I-TASSER/
单独注册账号,一次只能提交一个任务。
3 ab initio:QUARK
原理:1973年science,Anfinsen:蛋白质的三维结构决定于自身氨基酸序列,并且处于最低自由能状态。 QUARK适用于没有同源模板的蛋白质,且氨基酸序列长度应在200以内。 单独注册账号,一次只能提交一个任务。
https://zhanglab.ccmb.med.umich.edu/QUARK/
4 ensemble method:ROBETTA
原理:综合了前三种方法,将氨基酸序列分段,情况不同的片段采用不同的方法。
http://robetta.bakerlab.org/
模型质量评估
https://servicesn.mbi.ucla.edu/SAVES/
蛋白质三级结构的比对
http://superpose.wishartlab.com/
蛋白质三维结构比对就是对蛋白质三维空间结构的相似新进行比较。
-
可用于探索蛋白质进化及同源关系
-
改进序列比对的精度
-
改进蛋白质结构预测工具
-
为蛋白质结构分类提供依据
-
帮助了解蛋白质功能
结构比对的结果可以用很多种参数来衡量,最常用的是root mean squared deviations(RMSD)。如果两个结构的RMSD为0埃,则结构一致,可以完全重合;一般来说RMSD小于3埃时,认为两个结构相似。 蛋白结构叠合
https://spdbv.vital-it.ch/
蛋白质表面的分子性质
Guosen Zhao
Graduate Student in Plant Immunity
My research interests include Plant immunity, Bioinformatics and Running.